learn what commands to use when navigating the file system
time Time estimation: 30 minutes
Tutorial on the linux command line
We will first hold your hand: type over these commands below step by step, and watch what they do.
Use cd to change the current working directory (user bits). To create your own directories use the mkdir (make directory) command.
$ cd ~
$ mkdir sequences
$ cd sequences
$ mkdir proteins
$ cd proteins
$ pwd
/home/bits/sequences/proteins
$ cd ../..
$ pwd
/home/bits
To create a new file, use the touch command:
$ cd ~/sequences/proteins/
$ touch my_sequence.txt
$ ls -l
-rw-r--r-- 1 bits users 0 Sep 19 15:56 my_sequence.txt
In the last command above, the -l (a lowercase “L”, not a “1” (one)) option was used with the ls command. The -l indicates that you want the directory contents shown in the “long listing” format.
Most commands accept options. But which options can you use? The command man helps you. Type man followed by the command name. E.g. man ls to see what options are available for the ls command. You get a the list of options. Keep pressing Space until the page stops scrolling, then enter “q” to return to the command prompt.
Luckily, most tools have the –help option. (ls –help for example). These 2 methods should help you further. To see what options can be used with ls, enter man ls.
$ man ls
To delete a file, use the rm (remove) command:
$ cd ~/sequences/proteins/
$ ls
my_sequence.txt
$ rm my_sequence.txt
$ ls
$
To remove a directory, use the rmdir (remove directory) command. The directory needs to be empty to do this.
$ cd ~/sequences/
$ ls
proteins
$ rmdir proteins
$ ls
$
To copy a file, use the cp (copy) command:
$ cd ~/sequences
$ touch testfile1
$ ls
testfile1
$ cp testfile1 testfile2
$ ls
testfile1 testfile2
To rename a file, or to move it to another directory, use the mv (move) command:
$ cd
$ touch testfile3
$ mv testfile3 junk
$ mkdir testdir
$ mv junk testdir
$ ls testdir
junk
To download a file, use the wget command:
$ cd ~/Downloads
$ wget http://data.bits.vib.be/pub/trainingen/Linux/sample.sam
$ ls sample.sam
$
The commands covered so far represent a small but useful subset of the many commands available on a typical Linux system.
The ‘{‘ and ‘}’ can group arguments but you can also create the structure step by step.
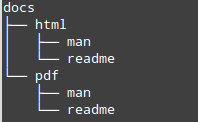
The little tree figure above is created with the ‘tree’ command. Display such a tree.
tree /home/bits/docs/
Downloading and storing bioinformatics data
Create a project folder
The first thing to do when you start a bioinformatics project, is to create a structure of folders to put your data in an organised fashion.
Downloading
As an example, we will download the rice genome from the Rice Annotation Project database. But first create the folder structure.
Create following folder structure.
$ mkdir "Rice Example"
$ cd Rice\ Example
$ mkdir Genome\ data
$ cd Genome\ data
$ mkdir Sequence
$ mkdir Annotation
$ cd
** Be aware of white spaces on the command line!**
On the command line, programs, options and arguments are separated by white spaces. If you choose to use a folder name containing a white space, it will interpret every word as an option or argument. So you have to tell Bash to ignore the white space. This can be done by: putting strings between quotes like ‘ or “ escape a white space with . See the examples above.
Hence, you might save yourself some trouble (and typing!) by putting _ instead of white spaces in names. Also make sure to use tab expansion, wherever possible!
Download the genome data directly on the command line
Download the genome data to the “Rice example”/”Genome data”/Sequence folder. Use wget to download from the link.
Right-click on the download link, and copy the download link. The download link is: http://rapdb.dna.affrc.go.jp/download/archive/build5/IRGSPb5.fa.masked.gz
Go the directory and execute wget
$ cd ## to go back to the home directory
$ cd Ric<tab>
$ cd Gen<tab>/Seq<tab>
$ wget http://rapdb.dna.affrc.go.jp/download/archive/build5/IRGSPb5.fa.masked.gz
--2013-10-15 09:36:01-- http://rapdb.dna.affrc.go.jp/download/archive/build5/IRGSPb5.fa.masked.gz
Resolving rapdb.dna.affrc.go.jp (rapdb.dna.affrc.go.jp)... 150.26.230.179
Connecting to rapdb.dna.affrc.go.jp (rapdb.dna.affrc.go.jp)|150.26.230.179|:80... connected.
HTTP request sent, awaiting response... 200 OK
Length: 122168025 (117M) [application/x-gzip]
Saving to: `IRGSPb5.fa.masked.gz'
100%[======================================>] 122,168,025 973K/s in 2m 40s
2013-10-15 09:38:42 (747 KB/s) - `IRGSP-1.0_genome.fasta.gz' saved [122168025/122168025]
$ ls
IRGSPb5.fa.masked.gz
Allright. We have fetched our first genome sequence!
Did your data get through correctly?
Large downloads or slow downloads like this can take a long time. Plenty of opportunity for the transfer to go wrong. Therefore, large downloads should always have a checksum mentioned. You can find the md5 checksum on the downloads page. The md5 checksum is an unique string identifying (and calculated from) this data. Once downloaded, you should calculate this string yourself with md5sum.
You should go to the rice genome download page, and compare this string with the MD5 checksum mentioned over there. You can do this manually. Now that you know the concept of checksums, there is an easier way to verify the data using md5sum. Can you find the easier way?
Search how to use md5sum to check the downloaded files with the .md5 file from the website. Check the man page
$ man md5sum
It does not say much: in the end it refers to
$ info coreutils 'md5sum invocation'
Reading the options, there is one option sounding promising:
`-c'
`--check'
Read file names and checksum information (not data) from each FILE
(or from stdin if no FILE was specified) and report whether the
checksums match the contents of the named files.
This way we can check the download:
$ wget http://rapdb.dna.affrc.go.jp/download/archive/build5/IRGSPb5.fa.masked.gz.md5
--2013-10-15 09:47:02-- http://rapdb.dna.affrc.go.jp/download/archive/build5/IRGSPb5.fa.masked.gz.md5
Resolving rapdb.dna.affrc.go.jp (rapdb.dna.affrc.go.jp)... 150.26.230.179
Connecting to rapdb.dna.affrc.go.jp (rapdb.dna.affrc.go.jp)|150.26.230.179|:80... connected.
HTTP request sent, awaiting response... 200 OK
Length: 55 [application/x-gzip]
Saving to: `IRGSPb5.fa.masked.gz.md5'
100%[======================================>] 55 --.-K/s in 0s
2013-10-15 09:47:03 (757 KB/s) - `IRGSPb5.fa.masked.gz.md5' saved [55/55]
$ ls
IRGSPb5.fa.masked.gz IRGSPb5.fa.masked.gz.md5
$ md5sum -c IRGSPb5.fa.masked.gz.md5
IRGSPb5.fa.masked.gz: OK
Ensuring integrity of downloads
A handy tool to use is the DownThemAll addon for Firefox, in which you have to provide the checksum at the time of download. It will automatically check whether the download is finished.
The Short Read Archive (SRA), storing NGS data sets, makes use of Aspera to download data a great speeds, ensuring integrity. To download from SRA using aspera in linux, follow the this guide from EBI.
Extracting the data
What type of file have you downloaded?
$ file IRGSPb5.fa.masked.gz
IRGSPb5.fa.masked.gz: gzip compressed data, was "IRGSPb5.fa.masked", from Unix, last modified: Wed Aug 18 03:45:47 2010
It is a compressed file. Files are compressed to save storage space. Before using these files, you have to decompress them. What can you do with this type of file? Check the command apropos.
$ apropos gzip
gzip (1) - compress or expand files
lz (1) - gunzips and shows a listing of a gzip'd tar'd archive
tgz (1) - makes a gzip'd tar archive
uz (1) - gunzips and extracts a gzip'd tar'd archive
zforce (1) - force a '.gz' extension on all gzip files
apropos is a command that helps you discover new commands. In case you have a type of file that you don’t know about, use apropos to search for corresponding programs.
Decompress the file. Check the man page of gzip. From the man page:<pre>gunzip [ -acfhlLnNrtvV ] [-S suffix] [ name … ]</pre>
$ gunzip IRGSPb5.fa.masked.gz
$ ls
IRGSPb5.fa.masked IRGSPb5.fa.masked.gz.md5
keypoints Key points
Where can I find command line information
What are useful commands
congratulations Congratulations on successfully completing this tutorial!